Le jeu de données est structuré sous forme de tableau où :
À partir de la description et de l’aperçu avec head(data), on observe :
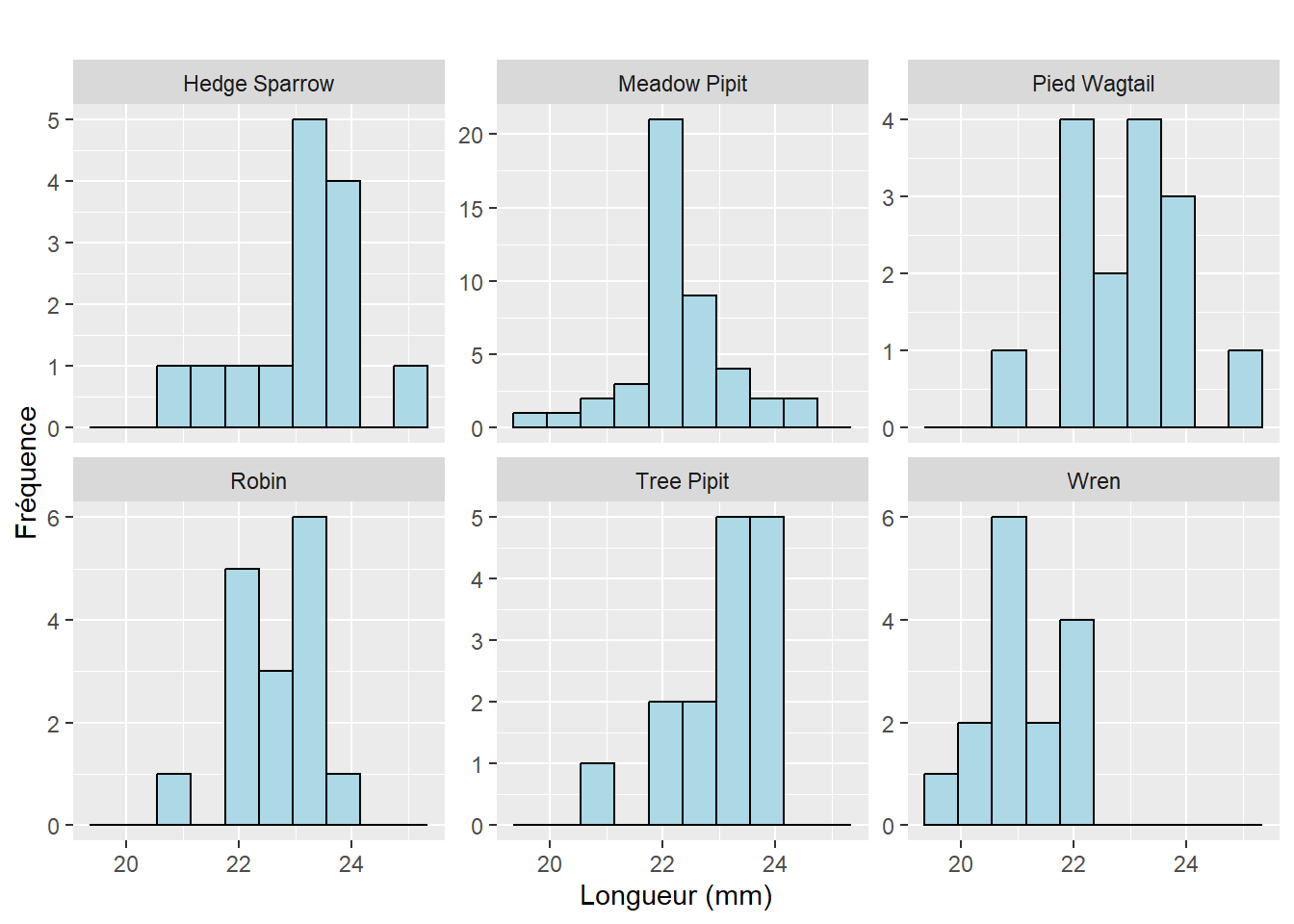
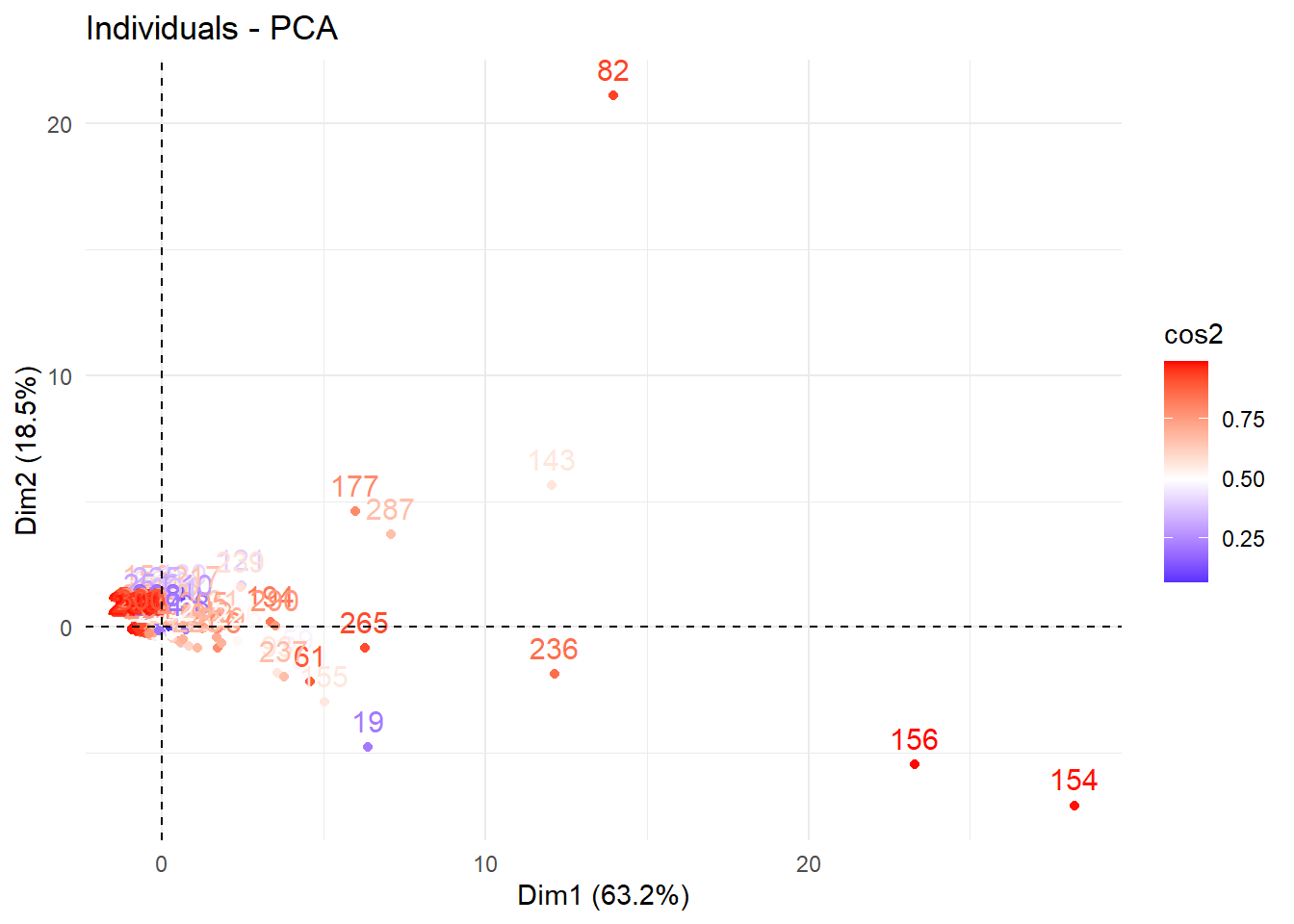
300 lignes, ce qui signifie que 300 gènes distincts sont analysés
13 colonnes au total, parmi lesquelles :
Le jeu de données contient donc 300 gènes (individus statistiques) décrits par 12 conditions biologiques (variables quantitatives).
Les variables quantitatives sont celles qui sont de type numérique, qui représentent des mesures continues, et peuvent être comparées par des opérations mathématiques (moyenne, variance, corrélation). Dans ce jeu de données, il s’agit des 12 colonnes évoquées plus haut :
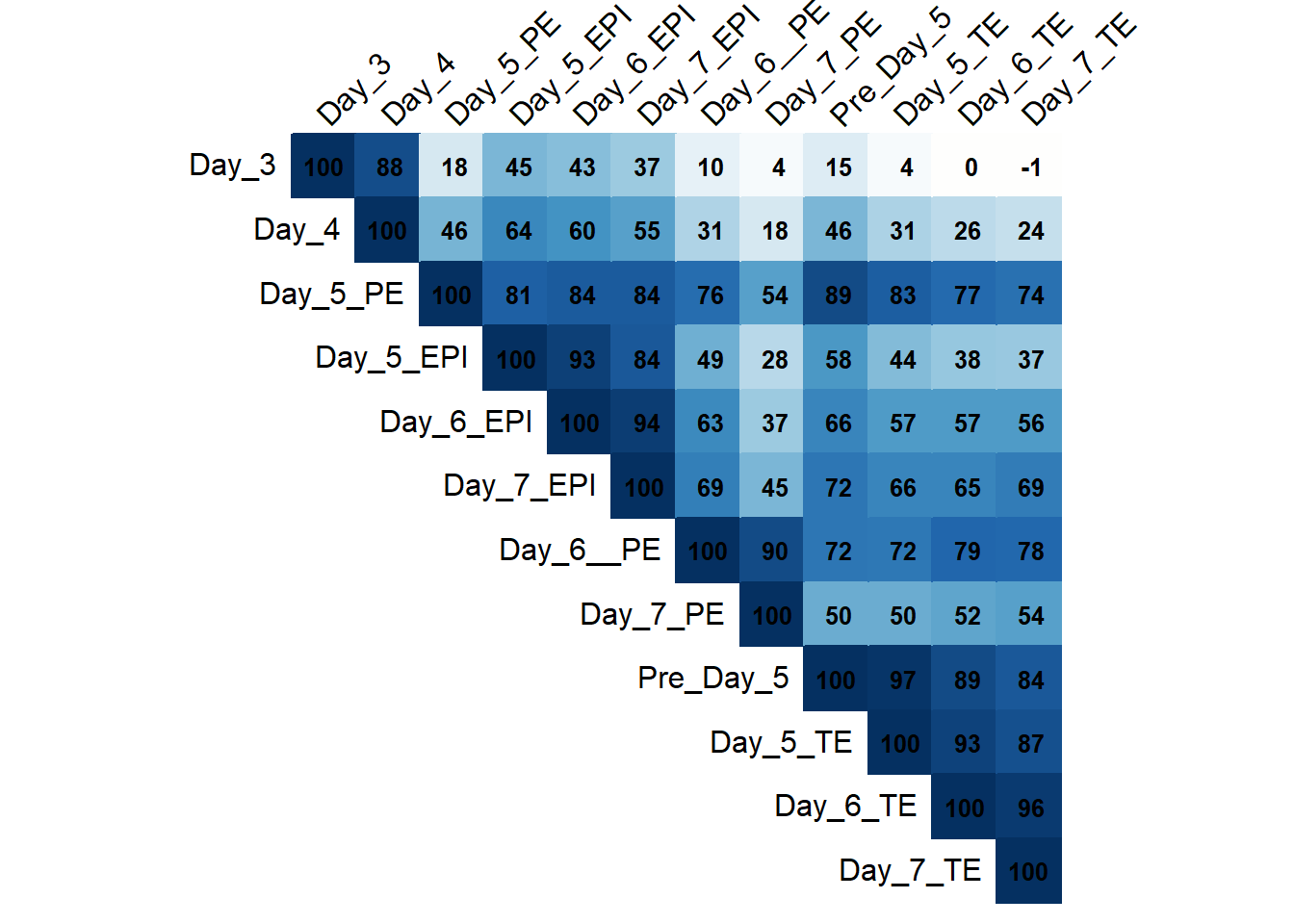
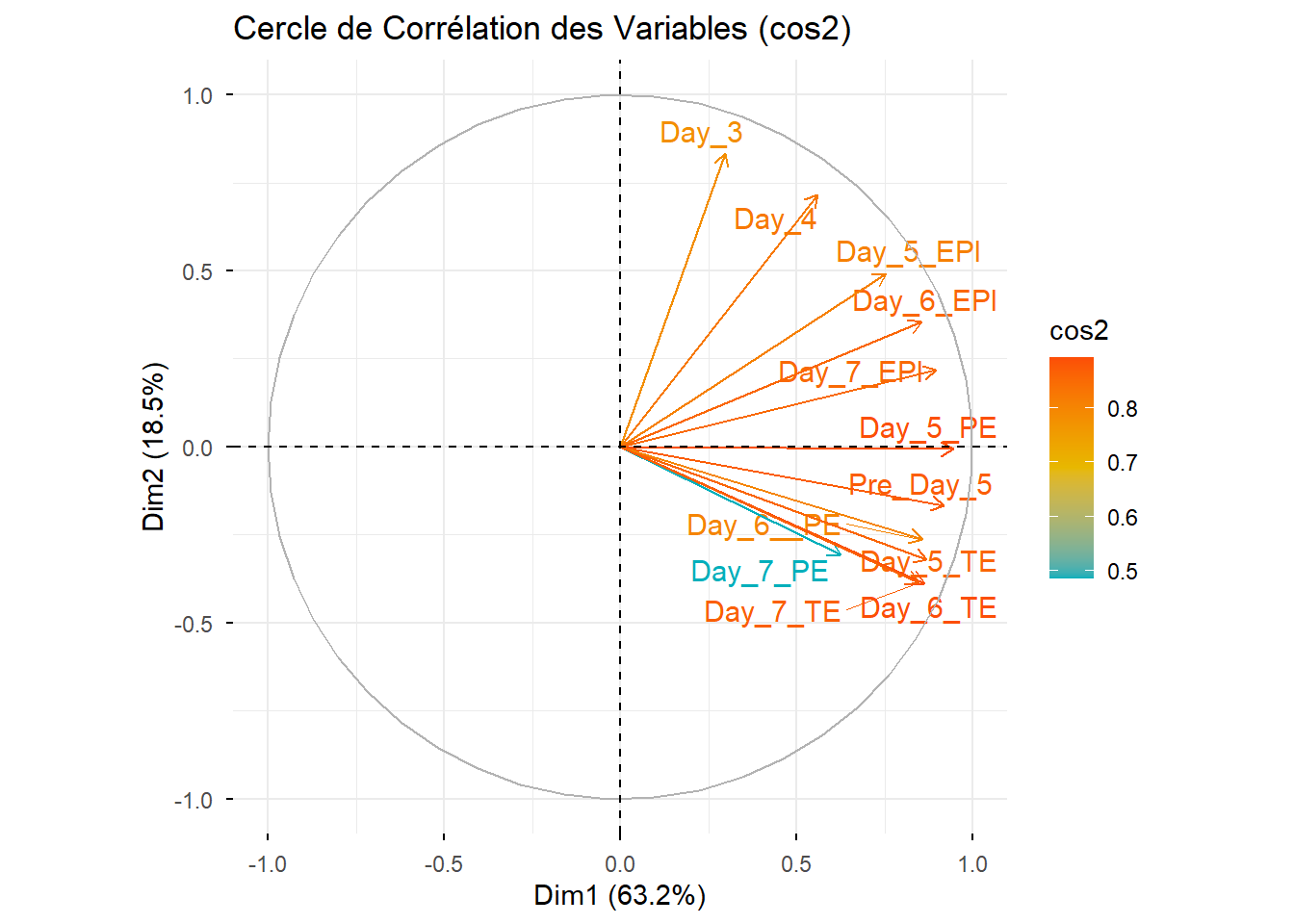
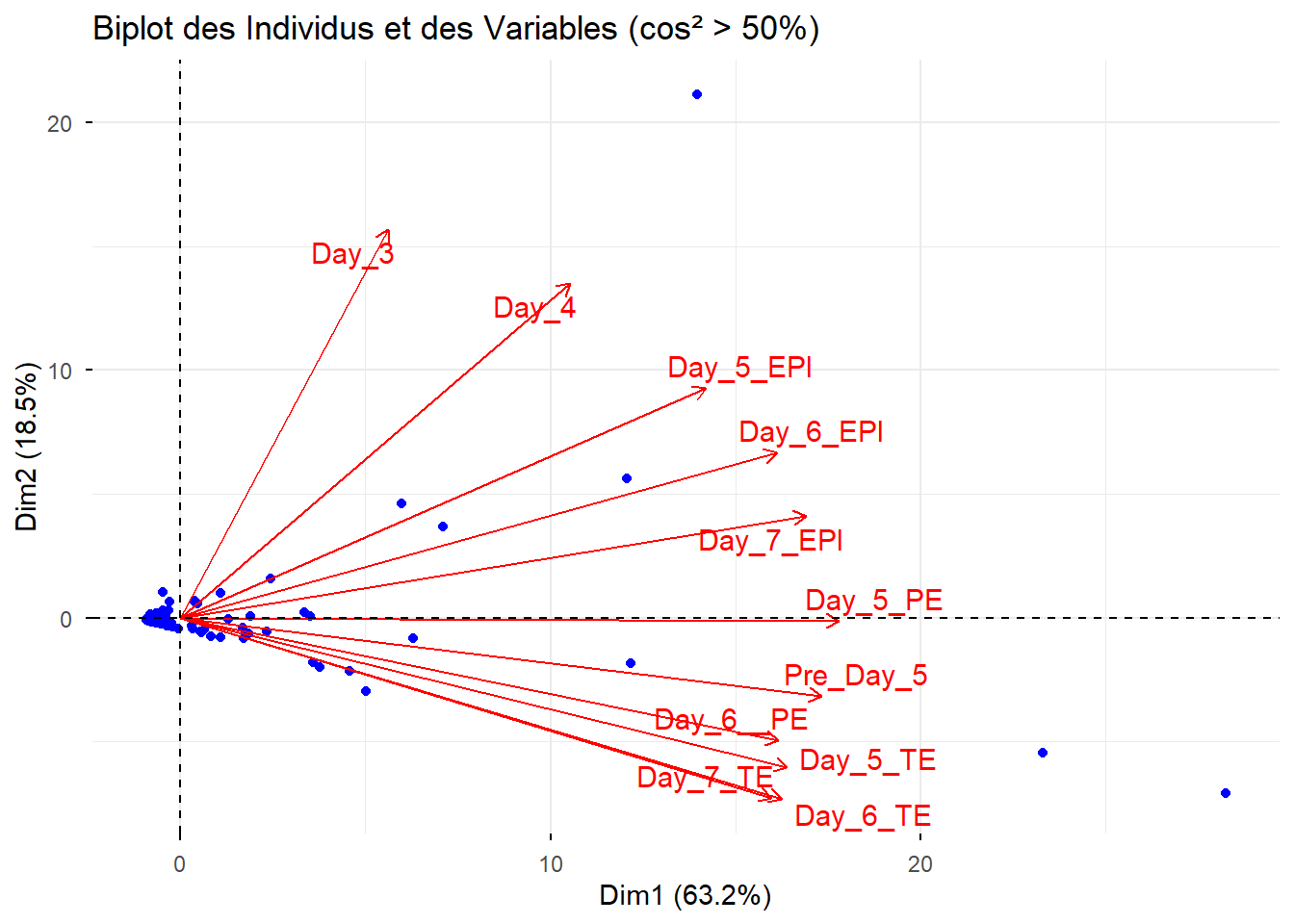
Day_3, Day_4, Pre_Day_5, Day_5_EPI, Day_5_PE, Day_5_TE, Day_6_EPI, Day_6_PE, Day_6_TE, Day_7_EPI, Day_7_PE et Day_7_TE.
Chaque valeur numérique correspond à un niveau d’expression génique mesuré pour un gène donné dans une condition biologique précise.
L’Analyse en Composantes Principales est une méthode algébrique. Elle repose sur :
Ces concepts mathématiques n’ont de sens que pour des variables quantitatives. Conséquences si cette distinction n’est pas faite, inclure une variable qualitative dans une ACP entraînerait une impossibilité mathématique ! Cette sélection garantit la validité statistique de l’analyse.
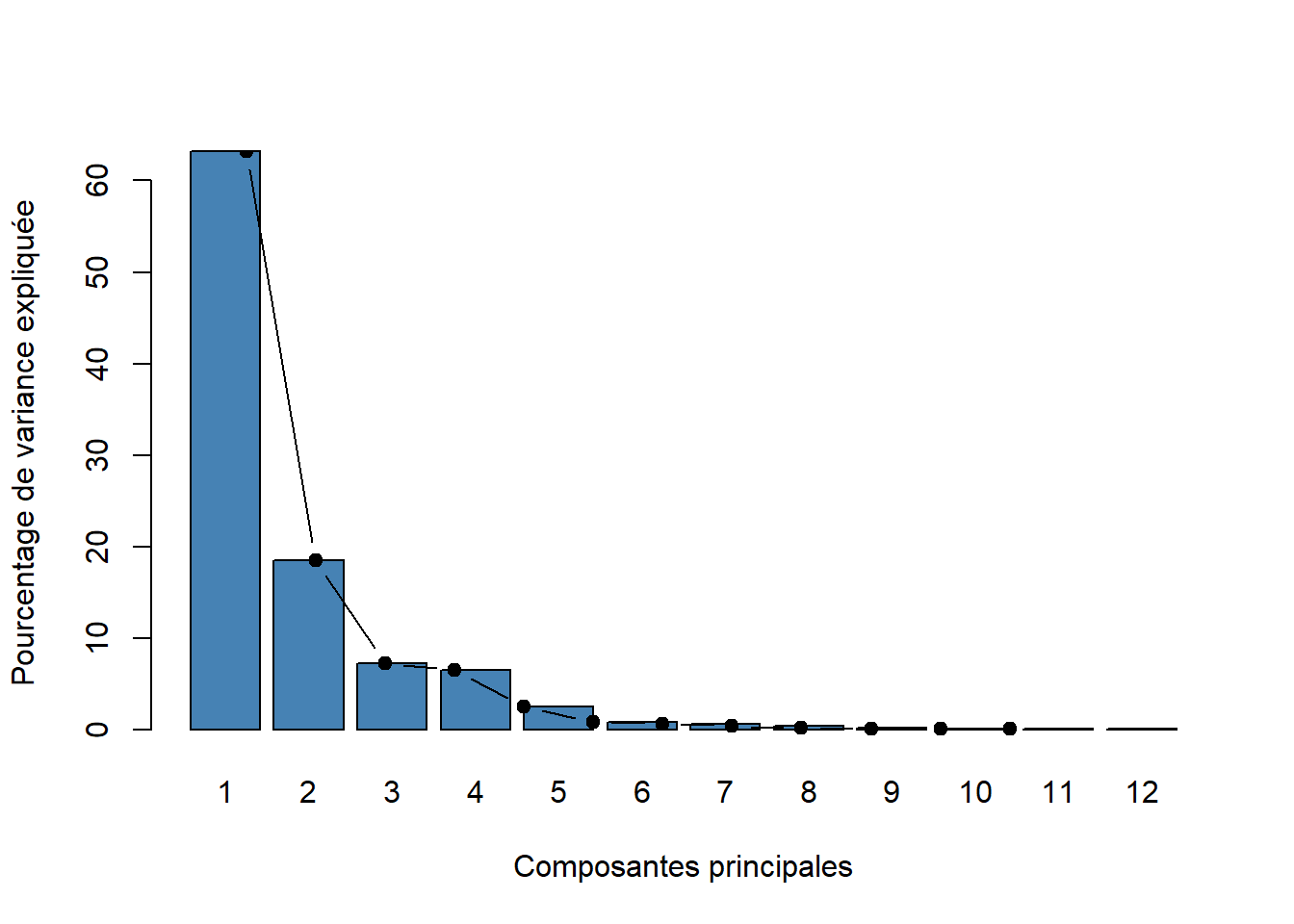
L’Analyse en Composantes Principales vise à résumer l’information contenue dans un grand nombre de variables corrélées par un petit nombre de variables non corrélées, appelées composantes principales. Plus précisément, cette analyse permet de :
Réduire la dimension des données
Conserver un maximum de variance
Identifier les structures dominantes
Faciliter la visualisation et l’interprétation de corrlations entre variables
Cette analyse est particulièrement adaptée ici car il y a un grand nombre de variables, les 12 conditions biologiques fortement dépendantes biologiquement (mêmes processus de développement). On s’attend donc à de fortes corrélations, ce qui est une situation idéale pour l’ACP. L’étude ne cherche pas à tester une hypothèse précise, mais à :
L’ACP est une méthode exploratoire non supervisée, parfaitement adaptée à cet objectif. Dans ce contexte, l’ACP doit permettre de :
- Identifier si les conditions se regroupent :
- Par jour de développement
- Par type cellulaire
- Comprendre quels ensembles de conditions :
- Partagent des profils d’expression similaires
- S’opposent sur certains axes